









Innhold
Bourneville tuberøs sklerose
Hva er det ?
Bourneville tuberøs sklerose er en kompleks genetisk sykdom karakterisert ved utvikling av en godartet (ikke-kreft) svulst i forskjellige deler av kroppen. Disse svulstene kan da lokaliseres i huden, hjernen, nyrene og andre organer og vev. Denne patologien kan også forårsake alvorlige problemer i utviklingen av individet. Imidlertid varierer de kliniske manifestasjonene og alvorlighetsgraden av sykdommen fra pasient til pasient.
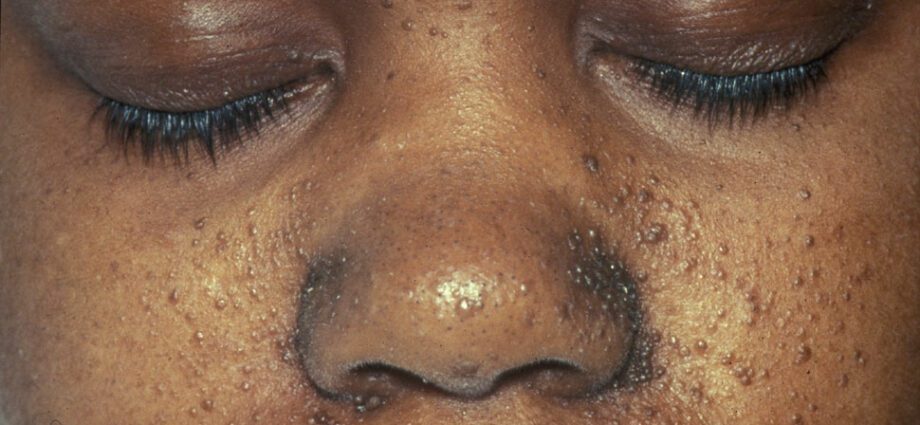
De tilknyttede hudavvikene ligner generelt på flekker på huden eller områder der huden er lysere enn på resten av kroppen. Utviklingen av svulster i ansiktet kalles angiofibrom.
I sammenheng med hjerneskade er de kliniske tegnene epileptiske anfall, atferdsproblemer (hyperaktivitet, aggressivitet, intellektuelle funksjonshemninger, lærevansker osv.). Noen barn med sykdommen har til og med en eller annen form for autisme, utviklingsforstyrrelser, som påvirker sosiale interaksjoner og kommunikasjon. Godartede hjernesvulster kan også forårsake komplikasjoner som kan være dødelige for personen.
Utviklingen av svulster i nyrene er vanlig hos personer med tuberøs sklerose. Dette kan forårsake alvorlige komplikasjoner i nyrefunksjonen. I tillegg kan det utvikle seg svulster i hjertet, lungene og netthinnen. (2)
Det er en sjelden sykdom, hvis prevalens (antall tilfeller i en gitt befolkning på et gitt tidspunkt) utgjør 1/8 til 000/1 personer. (15)
Symptomer
De kliniske manifestasjonene assosiert med tuberøs sklerose av Bourneville varierer i henhold til de berørte organene. I tillegg varierer symptomene forbundet med sykdommen mye fra individ til individ. Med symptomer som spenner fra milde til alvorlige.
De mest kjente symptomene på denne sykdommen inkluderer epileptiske anfall, kognitive og atferdsforstyrrelser, hudavvik osv. De organene som oftest rammes er: hjernen, hjertet, nyrene, lungene og huden.
Utvikling av ondartede (kreft)svulster er mulig ved denne sykdommen, men er sjeldne og påvirker hovedsakelig nyrene.
De kliniske tegnene på sykdommen i hjernen stammer fra angrep på forskjellige nivåer:
- skade på kortikale tuberkler;
– ependymale knuter (SEN);
– gigantiske ependymale astrocytomer.
De resulterer i: utvikling av mental retardasjon, lærevansker, atferdsforstyrrelser, aggressivitet, oppmerksomhetsforstyrrelser, hyperaktivitet, tvangslidelser, etc.
Nyreskade er preget av utvikling av cyster eller angiomyolipomer. Disse kan føre til nyresmerter og til og med nyresvikt. Hvis kraftig blødning er merkbar, kan det være fra alvorlig anemi eller høyt blodtrykk. Andre mer alvorlige, men sjeldne konsekvenser kan også være synlige, spesielt utviklingen av karsinomer (svulst i cellebestanddelene i epitelet).
Øyeskade kan ligne på synlige flekker på netthinnen, og forårsake synsforstyrrelser eller til og med blindhet.
Hudunormaliteter er mange:
– hypomelaniske makuler: som resulterer i lyse flekker på huden, hvor som helst på kroppen, som følge av mangel på melanin, et protein som gir farge til huden;
- utseendet av røde flekker i ansiktet;
- misfargede flekker på pannen;
– andre hudavvik, avhengig av individ til individ.
Lungelesjoner er tilstede hos 1/3 av pasientene med en liten kvinnelig overvekt. De tilhørende symptomene er da mer eller mindre alvorlige pustevansker.
Opprinnelsen til sykdommen
Opprinnelsen til sykdommen er genetisk og arvelig.
Overføring involverer mutasjoner i TSC1- og TSC2-genene. Disse genene av interesse spiller inn i dannelsen av proteiner: hamartin og tuberin. Disse to proteinene gjør det mulig, gjennom et interaktivt spill, å regulere celleproliferasjon.
Pasienter med sykdommen er født med minst én mutert kopi av disse genene i hver av sine celler. Disse mutasjonene begrenser da dannelsen av hamartin eller tubertin.
I sammenhengen hvor de to kopiene av genet er mutert, forhindrer de produksjonen av disse to proteinene fullstendig. Denne proteinmangelen tillater derfor ikke lenger kroppen å regulere veksten av visse celler og fører i denne forstand til utvikling av tumorceller i forskjellige vev og/eller organer.
Risikofaktorer
Risikofaktorene for å utvikle en slik patologi er genetiske.
Faktisk er overføringen av sykdommen effektiv gjennom en autosomal dominant modus. Enten er det muterte genet av interesse lokalisert ved et ikke-seksuelt kromosom. I tillegg er tilstedeværelsen av bare én av de to kopiene av det muterte genet tilstrekkelig for at sykdommen skal utvikle seg.
I denne forstand har en person som har en av disse to foreldrene som lider av sykdommen 50 % risiko for å utvikle den syke fenotypen selv.
Forebygging og behandling
Diagnosen av sykdommen er først og fremst differensiell. Den er basert på atypiske fysiske kriterier. I de fleste tilfeller er de første karakteristiske tegnene på sykdommen: tilstedeværelsen av tilbakevendende epileptiske anfall og forsinkelser i pasientens utvikling. I andre tilfeller resulterer disse første tegnene i hudflekker eller identifikasjon av en hjertesvulst.
Etter denne første diagnosen er ytterligere undersøkelser avgjørende for å validere diagnosen eller ikke. Disse inkluderer:
– en hjerneskanning;
– en MR (magnetisk resonansavbildning) av hjernen;
– en ultralyd av hjertet, leveren og nyrene.
Diagnosen kan være effektiv ved fødselen av barnet. Ellers er det viktig at det gjennomføres så raskt som mulig for å ta hånd om pasienten så raskt som mulig.
Foreløpig er det ingen kur mot sykdommen. De tilknyttede behandlingene er derfor uavhengige av symptomene som presenteres av hver enkelt.
Vanligvis gis antiepileptika for å begrense anfall. I tillegg er medisiner for behandling av tumorceller i hjernen og nyrene også foreskrevet. I sammenheng med atferdsproblemer er spesifikk behandling av barnet nødvendig.
Behandling av sykdommen er vanligvis langsiktig. (1)