









Innhold
Wilsons sykdom
Hva er det ?
Wilsons sykdom er en arvelig genetisk sykdom som forhindrer eliminering av kobber fra kroppen. Oppbygging av kobber i leveren og hjernen forårsaker lever- eller nevrologiske problemer. Prevalensen av Wilsons sykdom er svært lav, rundt 1 av 30 personer. (000) Det finnes en effektiv behandling for denne sykdommen, men den tidlige diagnosen er problematisk fordi den forblir stille i lang tid.
Symptomer
Kobberoppbygging begynner ved fødselen, men de første symptomene på Wilsons sykdom viser seg ofte ikke før i ungdomsårene eller i voksen alder. De kan være svært forskjellige fordi flere organer påvirkes av opphopning av kobber: hjertet, nyrene, øynene, blodet... De første tegnene er hepatiske eller nevrologiske i tre fjerdedeler av tilfellene (henholdsvis 40 % og 35 %), men de kan også være psykiatrisk, renal, hematologisk og endokrinologisk. Leveren og hjernen er spesielt påvirket fordi de allerede naturlig inneholder mest kobber. (2)
- Leversykdommer: gulsott, skrumplever, leversvikt ...
- Nevrologiske lidelser: depresjon, atferdsforstyrrelser, lærevansker, vanskeligheter med å uttrykke seg, skjelvinger, kramper og kontrakturer (dystoni) …
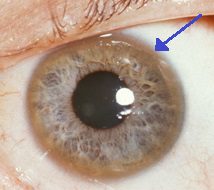
Keyser-Fleisher-ringen som omgir iris er karakteristisk for oppbygging av kobber i øyet. I tillegg til disse akutte symptomene kan Wilsons sykdom presentere med ukarakteristiske symptomer, som generell tretthet, magesmerter, oppkast og vekttap, anemi og leddsmerter.
Opprinnelsen til sykdommen
Ved opprinnelsen til Wilsons sykdom er det en mutasjon i ATP7B-genet lokalisert på kromosom 13, som er involvert i metabolismen av kobber. Det kontrollerer produksjonen av et ATPase 2-protein som spiller en rolle i transport av kobber fra leveren til andre deler av kroppen. Kobber er en nødvendig byggestein for mange cellefunksjoner, men i overkant av kobber blir det giftig og skader vev og organer.
Risikofaktorer
Overføring av Wilsons sykdom er autosomal recessiv. Det er derfor nødvendig å motta to kopier av det muterte genet (fra far og mor) for å utvikle sykdommen. Dette betyr at menn og kvinner er like utsatt og at to foreldre som bærer det muterte genet, men som ikke er syke, har en risiko i fire ved hver fødsel for å overføre sykdommen.
Forebygging og behandling
Det finnes en effektiv behandling for å stoppe utviklingen av sykdommen og redusere eller til og med eliminere symptomene. Det er også nødvendig at det initieres tidlig, men det tar ofte mange måneder etter symptomdebut å diagnostisere denne stille sykdommen, lite kjent og hvis symptomer peker på mange andre tilstander (hepatitt som er leverskade og depresjon for psykiatrisk involvering) .
En "chelaterende" behandling gjør det mulig å tiltrekke kobber og eliminere det i urinen, og dermed begrense dets akkumulering i organene. Den er basert på D-penicillamin eller Trientine, medisiner som tas gjennom munnen. De er effektive, men kan gi alvorlige bivirkninger (nyreskade, allergiske reaksjoner osv.). Når disse bivirkningene er for viktige, tyr vi til administrering av sink som vil begrense absorpsjonen av kobber i tarmene.
En levertransplantasjon kan være nødvendig når leveren er for skadet, noe som er tilfellet for 5 % av personer med Wilson sykdom (1).
En genetisk screeningtest tilbys til søsken til en berørt person. Det gir opphav til en effektiv forebyggende behandling i tilfelle det oppdages en genetisk abnormitet i ATP7B-genet.