









Innhold
Huntingtons sykdom
Hva er det ?
Huntingtons sykdom er en genetisk og arvelig nevrodegenerativ sykdom. Ved å ødelegge nevroner i visse områder av hjernen, forårsaker det alvorlige motoriske og psykiatriske lidelser og kan føre til fullstendig tap av autonomi og død. Genet hvis endring forårsaker sykdommen ble identifisert på 90-tallet, men Huntingtons sykdom er fortsatt uhelbredelig frem til i dag. Det rammer én av ti personer i Frankrike, som representerer rundt 10 pasienter.
Symptomer
Det kalles fortsatt noen ganger "Huntingtons chorea" fordi det mest karakteristiske symptomet på sykdommen er de ufrivillige bevegelsene (kalt choreiske) den forårsaker. Noen pasienter har imidlertid ikke choreiske lidelser, og symptomene på sykdommen er bredere: til disse psykomotoriske lidelsene kommer ofte psykiatriske og atferdsmessige lidelser. Disse psykiatriske lidelsene som forekommer hyppig ved sykdomsutbruddet (og noen ganger vises før de motoriske lidelsene) kan føre til demens og selvmord. Symptomer vises vanligvis rundt 40-50 års alder, men tidlige og sene former for sykdommen observeres. Merk at alle bærere av det muterte genet en dag erklærer sykdommen.
Opprinnelsen til sykdommen
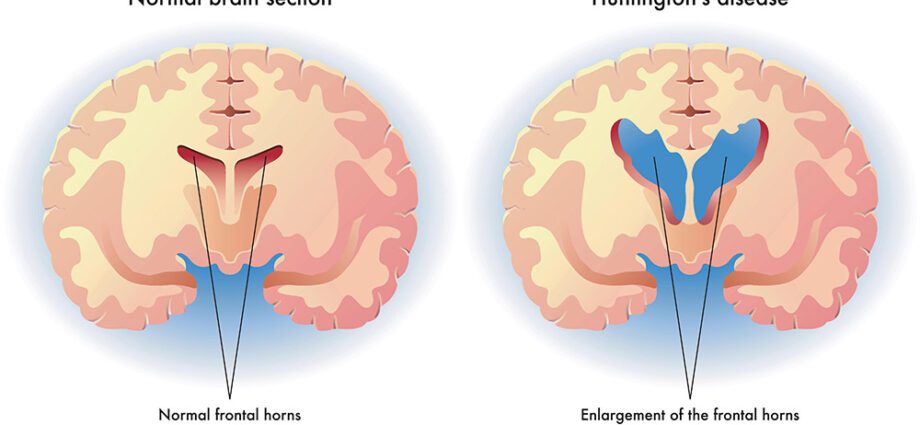
Den amerikanske legen George Huntington beskrev Huntingtons sykdom i 1872, men det var ikke før i 1993 at det ansvarlige genet ble identifisert. Det ble lokalisert på den korte armen til kromosom 4 og navngitt IT15. Sykdommen er forårsaket av en mutasjon i dette genet som styrer produksjonen av huntingtin-proteinet. Den nøyaktige funksjonen til dette proteinet er fortsatt ukjent, men vi vet at den genetiske mutasjonen gjør det giftig: det forårsaker avleiringer i midten av hjernen, mer presist i kjernen til nevroner i caudatkjernen, deretter i hjernebarken. Det skal imidlertid bemerkes at Huntingtons sykdom ikke er systematisk knyttet til IT15 og kan være forårsaket av mutering av andre gener. (1)
Risikofaktorer
Huntingtons sykdom kan overføres fra generasjon til generasjon (den kalles "autosomal dominant") og risikoen for overføring til avkommet er én av to.
Forebygging og behandling
Den genetiske screeningen av sykdommen hos personer i faresonen (med en familiehistorie) er mulig, men veldig overvåket av medisinsk profesjon, fordi resultatet av testen ikke er uten psykologiske konsekvenser.
Prenatal diagnose er også mulig, men det er strengt innrammet av loven, fordi det reiser spørsmål om bioetikk. En mor som vurderer en frivillig avbrytelse av svangerskapet i tilfelle at fosteret bærer det endrede genet har imidlertid rett til å be om denne prenatale diagnosen.
Til dags dato finnes det ingen kurativ behandling og kun behandling av symptomer kan lindre den syke og bremse den fysiske og psykiske forverringen: psykotrope stoffer for å lindre de psykiatriske lidelsene og episodene med depresjon som ofte går hånd i hånd med sykdommen. ; nevroleptika for å redusere choreiske bevegelser; rehabilitering gjennom fysioterapi og logopedi.
Jakten på fremtidige terapier er rettet mot transplantasjon av føtale nevroner for å stabilisere de motoriske funksjonene i hjernen. I 2008 beviste forskere fra Pasteur Institute og CNRS hjernens evne til å reparere seg selv ved å identifisere en ny kilde til nevronproduksjon. Denne oppdagelsen vekker nye forhåpninger for behandling av Huntingtons sykdom og andre nevrodegenerative tilstander, som Parkinsons sykdom. (2)
Genterapiforsøk pågår også i flere land og går i flere retninger, hvorav en er å blokkere uttrykket av det muterte huntingtin-genet.