









Innhold
talassemi
Thalassemia er et sett med arvelige blodsykdommer som påvirker produksjonen av hemoglobin (proteinet som er ansvarlig for transport av oksygen). De varierer i alvorlighetsgrad: noen forårsaker ingen symptomer mens andre er livstruende. En benmargstransplantasjon vurderes i de mest alvorlige tilfellene.
Thalassemi, hva er det?
Definisjon av talassemi
Thalassemi er preget av en defekt i produksjonen av hemoglobin. Som en påminnelse er hemoglobin et stort protein som finnes i røde blodceller (røde blodlegemer) hvis rolle er å sikre transporten av dixoygen fra luftveiene til resten av kroppen.
Det sies at talassemi er en sykdom i blodet. Røde blodlegemers transportfunksjon er svekket, noe som kan få alvorlige konsekvenser for kroppen. På dette tidspunktet er det viktig å merke seg at det er flere typer thalassemi som ikke har samme egenskaper eller samme alvorlighetsgrad. Noen har ingen symptomer mens andre er livstruende.
Årsaker til talassemi
Thalassemia er genetiske sykdommer. De skyldes endringen av ett eller flere gener involvert i syntesen av hemoglobin, og mer nøyaktig endringen av genene som er involvert i produksjonen av hemoglobinproteinkjeder. Det er fire av disse: to alfa-kjeder og to beta-kjeder.
Hver av disse kjedene kan påvirkes ved thalassemi. Vi kan også skille mellom:
- alfa-thalassemi karakterisert ved en endring av alfa-kjeden;
- beta-thalassemi karakterisert ved en endring av beta-kjeden.
Alfa-thalassemia og beta-thalassemia avhenger av antallet gener som er endret. Jo viktigere det er, desto større er alvorlighetsgraden.
Diagnose av talassemi
Diagnosen thalassemi stilles ved blodprøve. Den fullstendige blodtellingen gjør det mulig å vurdere utseendet og antallet røde blodlegemer, og dermed vite den totale mengden hemoglobin. Biokjemiske analyser av hemoglobin gjør det mulig å skille alfa-thalassemia fra beta-thalassemia. Til slutt gjør genetiske analyser det mulig å evaluere antall endrede gener og dermed definere alvorlighetsgraden av talassemi.
Berørte personer
Thalassemia er arvelige genetiske sykdommer, det vil si overføres fra foreldre til barn. De når hovedsakelig mennesker fra middelhavskanten, Midtøsten, Asia og Afrika sør for Sahara.
I Frankrike er forekomsten av alfa-thalassemi estimert til 1 av 350 personer. Forekomsten av beta-thalassemi er estimert til 000 fødsler per 1 per år over hele verden.
Symptomer på talassemi
Symptomene på talassemi varierer sterkt fra tilfelle til tilfelle, og avhenger hovedsakelig av graden av endring av genene som er involvert i produksjonen av hemoglobinproteinkjeder. Thalassemia kan være symptomfri i sine mindre former og være livstruende i sine mer alvorlige former.
Symptomene nevnt nedenfor gjelder kun mellomliggende til store former for talassemi. Dette er bare hovedsymptomene. Svært spesifikke symptomer kan noen ganger sees avhengig av typen thalassemi.
Anemi
Det typiske tegnet på talassemi er anemi. Dette er mangel på hemoglobin som kan resultere i forskjellige symptomer:
- tretthet;
- kortpustethet;
- blekhet;
- ubehag;
- hjertebank.
Intensiteten av disse symptomene varierer avhengig av alvorlighetsgraden av talassemien.
Gulsott
Personer med talassemi kan ha gulsott (gulsott) som er synlig på huden eller det hvite i øynene.
gallestein
Steindannelse inne i galleblæren kan også sees. Beregninger er som "små rullesteiner".
splenomegali
Splenomegali er en forstørrelse av milten. En av rollene til dette organet er å filtrere blodet og filtrere skadelige stoffer, inkludert unormale røde blodlegemer. Ved talassemi er milten sterkt mobilisert og øker gradvis i størrelse. Smerte kan føles.
Andre, sjeldnere symptomer
Mer sjelden kan alvorlige former for talassemi føre til andre abnormiteter. For eksempel kan det observeres:
- hepatomegali, det vil si en økning i leverens størrelse;
- bein misdannelser;
- forsinket barns utvikling;
- magesår.
Behandling av talassemi er avgjørende for å begrense forekomsten av disse komplikasjonene.
Behandlinger for talassemi
Behandlingen av thalassemi avhenger av mange parametere, inkludert typen thalassemi, alvorlighetsgraden og tilstanden til den berørte personen. De mest mindre formene krever ikke behandling, mens de alvorlige formene krever svært regelmessig medisinsk overvåking.
Behandlingene nevnt nedenfor gjelder kun mellomliggende til store former for talassemi
Korrigering av anemi
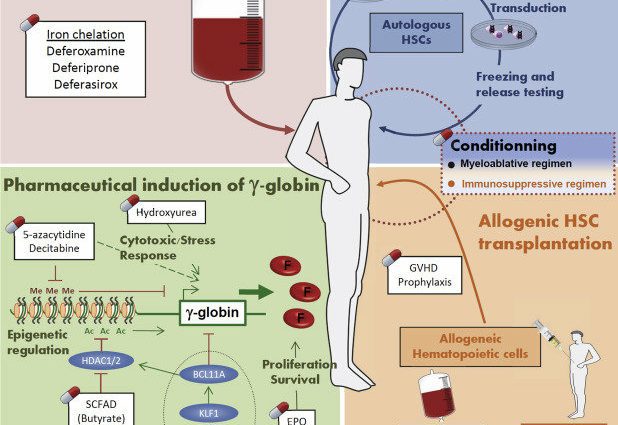
Når mangelen på hemoglobin er for stor, er regelmessige blodoverføringer nødvendig. De innebærer å injisere den berørte personen med blod eller røde blodceller tatt fra en giver for å opprettholde et akseptabelt nivå av røde blodlegemer i blodet.
Vitamin B9 tilskudd
Det kan anbefales å starte daglig vitamin B9-tilskudd fordi behovet for dette vitaminet øker ved thalassemi. Vitamin B9 er involvert i produksjonen av røde blodlegemer.
splenektomi
En splenektomi er kirurgisk fjerning av milten. Denne operasjonen kan vurderes når anemien er svært viktig.
Behandling av jernoverskudd
Personer med talassemi har en overbelastning av jern i kroppen. Denne opphopningen kan føre til forskjellige komplikasjoner. Dette er grunnen til at jernkelatorer tilbys for å fjerne overflødig jern.
Beinmargstransplantasjon
En benmargstransplantasjon er den eneste behandlingen som kan kurere thalassemi permanent. Dette er en tung behandling som kun tilbys ved de mest alvorlige formene av sykdommen.
Forhindre talassemi
Thalassemi er en arvelig genetisk sykdom. Det er ingen forebyggende tiltak.
På den annen side gjør genetiske tester det mulig å oppdage friske bærere (personer som har ett eller flere endrede gen(er), men som ikke er syke). Et par friske bærere bør informeres om risikoen for å føde et barn med thalassemi. I noen tilfeller kan denne risikoen vurderes av en genetiker. Prenatal diagnose kan også vurderes under visse forhold. Det bør diskuteres med legen din.